Wonpil Im, a professor in Lehigh’s Department of Biological Sciences and Bioengineering Department
Developed by Wonpil Im, a professor in Lehigh’s Department of Biological Sciences and Bioengineering Department, CHARMM-GUI (GUI = graphical user interface) is a program that simulates complex biomolecular systems simply, precisely and quickly. Im describes it as a “computational microscope” that enables scientists to understand molecular-level interactions that cannot be observed any other way. More information about CHARMM-GUI can be found in this video.
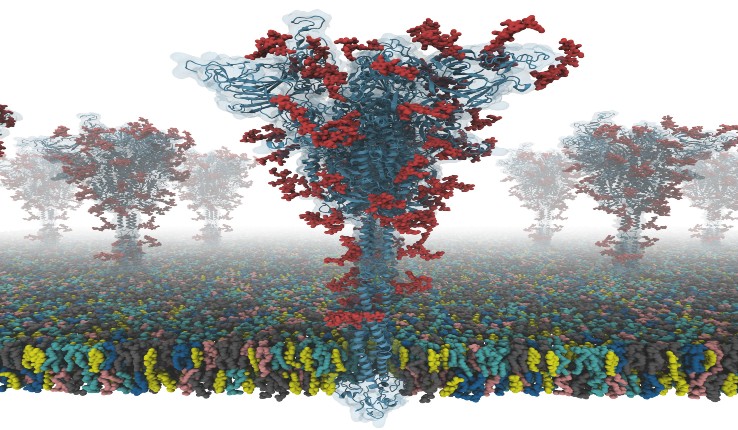
“Our models are the first fully-glycosylated full-length SARS-CoV-2 spike (S) protein models that are available to other scientists,” says Im. “I was fortunate to collaborate with Dr. Chaok Seok from Seoul National University in Korea and Dr. Tristan Croll from University of Cambridge in the U.K. Our team spent days and nights to build these models very carefully from the known cryo-EM structure portions. Modeling was very challenging because there were many regions where simple modeling failed to provide high-quality models.”
Scientists can use the models to conduct innovative and novel simulation research for the prevention and treatment of COVID-19, according to Im.
The S protein structure was determined with cryo-EM with the RBD up (PDB ID: 6VSB), and with the RBD down (PDB ID: 6VXX). But, this model has many missing residues. So, they first modeled the missing amino acid residues, and then other missing domains. In addition, they modeled all potential glycans (or carbohydrates) attached to the S protein. The glycans prevent antibody recognition, which makes it difficult to develop a vaccine. They also built a viral membrane system of an S protein for molecular dynamics simulation.
The team recommends reading the preprint paper, “Modeling and Simulation of a Fully-glycosylated Full-length SARS-CoV-2 Spike Protein in a Viral Membrane,” before using any of the models.